In 2012 the European Cardiovascular Research Institute (ECRI) was founded as a European collaborative effort to perform Investigator Sponsored Studies in the field of cardiology.

Talent - Study design
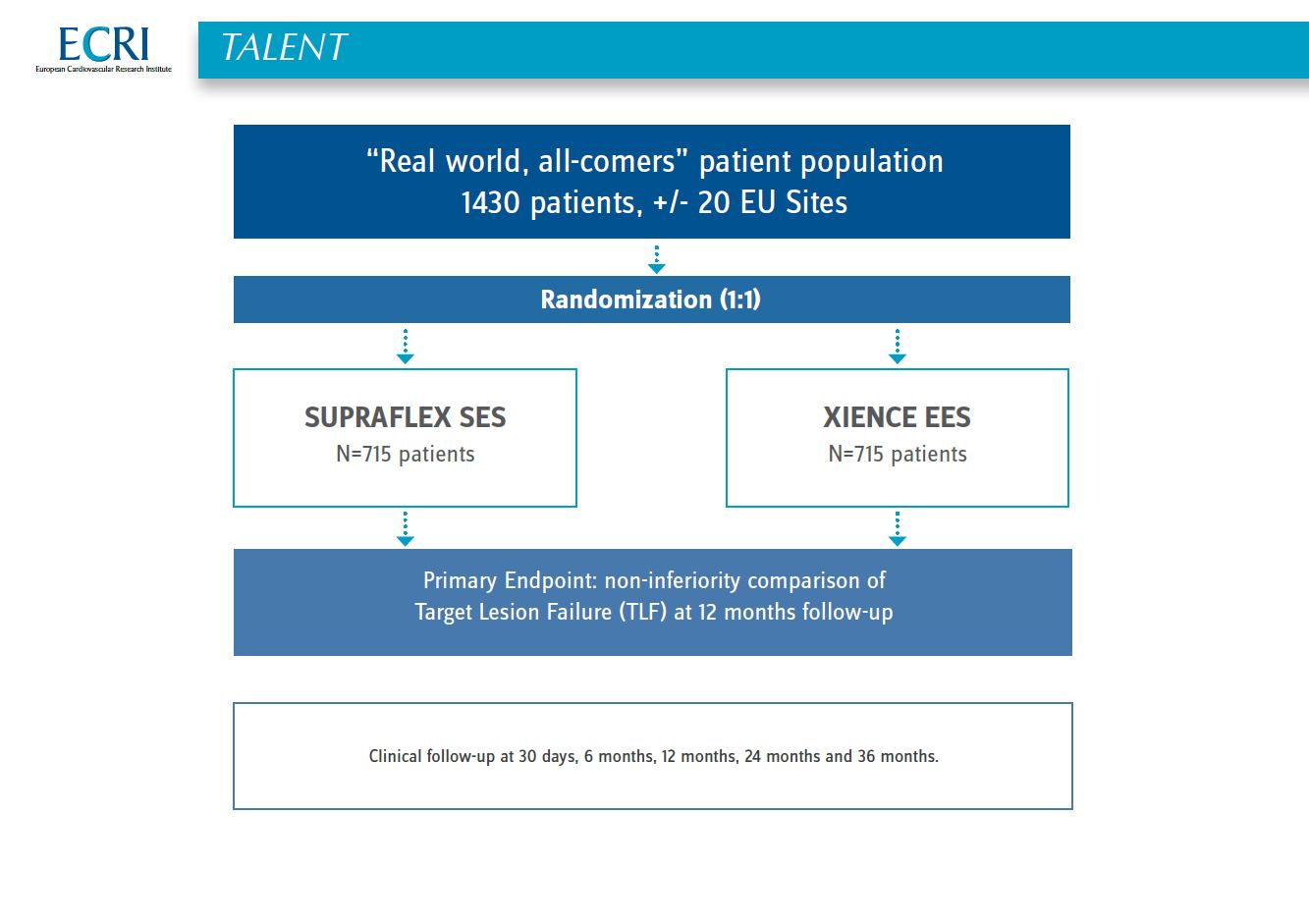
This is a prospective, randomized, 1:1 balanced, controlled, single-blind, multi-center study comparing clinical outcomes at 12 months between SUPRAFLEX and XIENCE in a “Real world, all comers” patient population (patients with symptomatic coronary artery disease including patients with chronic stable angina, silent ischemia, and acute coronary syndromes, who qualify for percutaneous coronary interventions).
1,430 patients will be enrolled to receive treatment with either the study device (SUPRAFLEX) or a control device (XIENCE).
All patients will be (at minimum) contacted at 30 days, 6 months, and 12 months post procedure to assess clinical status and adverse events. The 30 day and 12 month will be a clinic visit. All patients will have annual contact through 3 years follow-up to assess clinical status and adverse events.
The primary endpoint for this trial is a non-inferiority comparison of the device-oriented composite endpoint Target Lesion Failure (TLF) of the SUPRAFLEX group to the XIENCE group at 12 months post-procedure. TLF is a composite of clinical endpoint of
- cardiac death,
- myocardial infarction* not clearly attributable to a nontarget vessel and
- clinically-indicated target lesion revascularization (TLR).
*SCAI consensus for peri-procedure MI ≤48 hours, and 3rd universal definition for MI >48 hours after index procedure.